
Yuan Lab
- About
- Research
- People
- Publications
Our laboratory is a translational research lab studying pulmonary hypertension (PH), right heart failure and lung vascular (dys)function. The lab is specifically interested in examining physiological and pathophysiological roles of ion channels, membrane receptors and intracellular Ca2+ signaling in the pulmonary vasculature (including smooth muscle and endothelial cells) using combined techniques of patch clamp, digital imaging fluorescence microscopy and conventional molecular biological approaches.
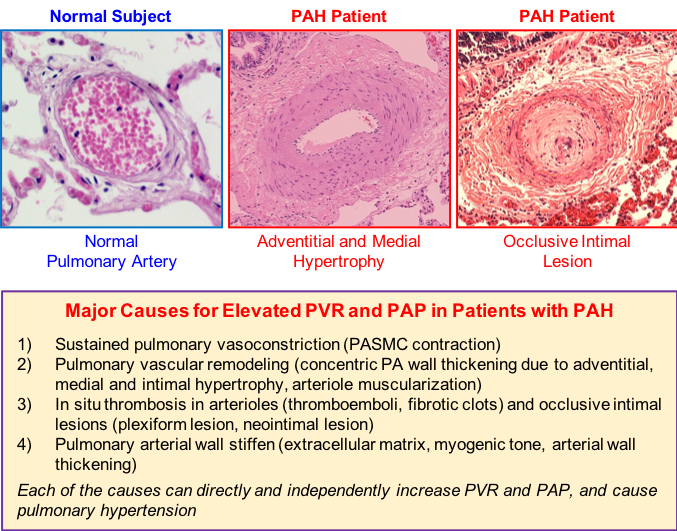
Figure 1. Major causes for the elevated pulmonary vascular resistance (PVR) and pulmonary arterial pressure (PAP) in patients with pulmonary arterial hypertension (PAH). Upper panels: Lung histology showing a small pulmonary artery (PA) from a normal subject (left), the remodeled or thickened PA from a PAH patient (middle), and the occluded PA from a PAH patient (right). Histological cross sections of small arteries in the lungs reveal the extensive hypertrophy of the adventitia, media and intima of PA (or concentric pulmonary arterial wall thickening) and occlusion of small PA that occur in PAH patients. Lower panel: List of the major causes leading to an increase in PVR in patients with PAH and animals with experimental pulmonary hypertension.
Idiopathic pulmonary arterial hypertension (PAH) is a progressive and fatal disease that predominantly affects women. Elevated pulmonary vascular resistance (PVR) in PAH patients results in an increase in afterload in the right ventricle (RV), leading to right ventricular hypertrophy, right heart failure, and death. Regardless of the initial genetic or pathogenic trigger, the pathogenesis of PAH can be attributed to sustained pulmonary vasoconstriction, concentric pulmonary vascular remodeling, in situthrombosis, and pulmonary vascular wall stiffening that directly increase PVR in patients with PAH (Figure 1). Understanding the pathogenic mechanisms of PAH is important for developing novel and effective therapeutic approaches for the disease.
The thickness and tissue mass of the pulmonary artery (PA) wall are maintained at an optimal level by a fine balance between cell proliferation and apoptosis. If there is more PASMC proliferation, the wall thickens, narrowing the lumen and ultimately leading to vascular narrowing and obliteration. The structural change leading to the pathological abnormality in the pulmonary vasculature is referred to as vascular remodeling. Both vasoconstriction and vascular remodeling also decrease vascular compliance (which, via distention and recruitment, normally accommodates increases in pulmonary arterial flow or cardiac output (CO), and increase PVR and PAP.
An increase in cytosolic free Ca2+ concentration ([Ca2+]cyt) in PASMC is not only a major trigger for pulmonary vasoconstriction but also an important stimulus for PASMC proliferation and migration. Intracellular Ca2+ is a critical second messenger responsible for linking external stimuli to contraction, migration, proliferation, and gene expression. A rise in [Ca2+]cyt rapidly increases nuclear [Ca2+] ([Ca2+]n). The elevated [Ca2+]cyt and [Ca2+]n both contribute to promoting proliferation by stimulating quiescent cells to enter the cell cycle (G0àG1 phase) and by driving proliferating cells through the cell cycle (G1àS phase and G2àM phase) and mitosis. In addition, elevated [Ca2+]cyt, by activating kinases (e.g., CaMK, MAPK) and transcription factors (e.g., NFAT, CREB, AP-1, NF-κB), promotes transcription of genes that are necessary for cell growth (Figure 2). A rise in [Ca2+]cyt and Ca2+/CaM-mediated activation of CaMK can also inactivate co-repressors associated with transcription factors and potentiate gene transcription. These results indicate that an increase in [Ca2+]cyt in PASMC, due to upregulated expression and/or increased activity of Ca2+-permeable channels, can serve as a shared mechanism for sustained pulmonary vasoconstriction and severe pulmonary vascular remodeling.
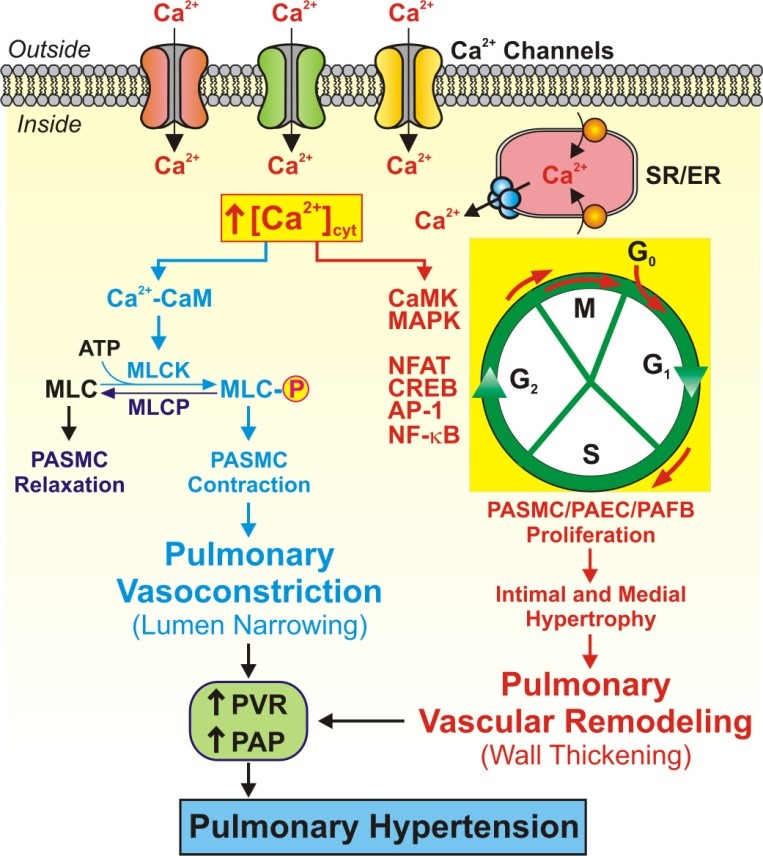
Figure 2. An increase in [Ca2+]cyt in PASMC triggers pulmonary vasoconstriction and stimulate PASMC proliferation and vascular remodeling. When [Ca2+]cyt rises due to Ca2+ influx through different Ca2+ channels in the plasma membrane and Ca2+ mobilization from the intracellular stores (e.g., SR/ER), Ca2+ binds calmodulin (CaM) which causes PASMC contraction by activating (via phosphorylation) myosin light chain kinase (MLCK). Increased [Ca2+]cyt also activates CaMK, MAPK and AKT, as well as other transcription factors (NFAT, CREB, AP-1 and NF-κB), to stimulate PASMC proliferation by propelling Ca2+-sensitive steps in the cell cycle progression.
The calcium-sensing receptor (CaSR) is a G protein-coupled receptor (GPCR) activated by extracellular Ca2+, polyamines (e.g., spermine), amino acids and neomycin. Activation of CaSR sets into motion a complex series of Ca2+ signaling events. CaSR activation increases the synthesis of inositol 1,4,5 trisphosphate (IP3) and diacylglycerol (DAG) via phospholipase C (PLC). IP3 binds to the IP3 receptor (IP3R) on the sarcoplasmic reticulum (SR) membrane and releases Ca2+ from the SR to the cytosol. Depletion of Ca2+ from the SR of PASMC induces Ca2+ entry through store-operated Ca2+ channels (SOC), also referred to as store-operated Ca2+ entry (SOCE) or capacitative Ca2+entry. DAG directly activates receptor-operated Ca2+ channels (ROC) in the plasma membrane; the Ca2+ entry through ROC is termed receptor-operated Ca2+ entry (ROCE). In addition to increasing [Ca2+]cytvia ROCE and SOCE, extracellular Ca2+-induced activation of CaSR also can activate other signal transduction pathways (e.g., Akt/mTOR) to induce cell proliferation (Figure 3).
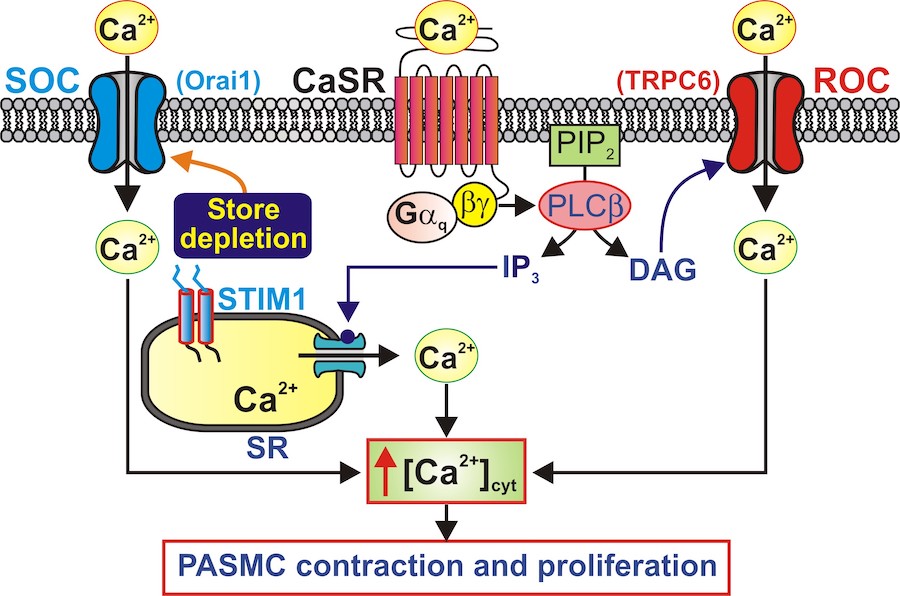
Figure 3. Putative role of CaSR in raising [Ca2+]cyt via Ca2+ release, ROCE through ROC (e.g., TRPC6) and SOCE through SOC (e.g., Orai1/STIM1) in PASMC.
We have previously shown that the extracellular Ca2+-induced increase in [Ca2+]cyt by activation of CaSR is enhanced and CaSR protein is markedly upregulated in IPAH-PASMC compared to normal PASMC. The inhibition of CaSR by siRNA in IPAH-PASMC attenuated the extracellular Ca2+-induced rise in [Ca2+]cyt and markedly inhibited IPAH-PASMC proliferation. Similarly, extracellular Ca2+-induced increase in [Ca2+]cyt by activation of CaSR is enhanced and CaSR mRNA and protein are highly upregulated in PASMC isolated from rats with monocrotaline (MCT)-induced PH (MCT-PH) compared to PASMC from control rats. Blockade of CaSR by the calcilytic NPS-2143 inhibited the development of MCT-PH.
Based on these results, we hypothesize that Ca2+ acts as an extracellular ligand as well as an intracellular signaling element to induce pulmonary vasoconstriction and vascular remodeling; upregulation of CaSR contributes to the enhanced Ca2+ influx (via, partially, its interaction with TRPC6 channels) and augmented PASMC proliferation in IPAH, whereas inhibition of CaSR is potentially a novel therapeutic approach for PH. Our long-term goals are to define the mechanisms underlying the Ca2+ signaling involved in enhanced PASMC proliferation and to explore CaSR as a potential therapeutic target for the treatment of IPAH.
Activity of voltage-gated K+ (Kv) channels in PASMC contributes to the control of membrane potential (Em). Downregulation of Kv channel expression and inhibition of Kv channel function cause membrane depolarization that subsequently opens VDCC, enhances Ca2+ influx, increases [Ca2+]cyt, causes pulmonary vasoconstriction and stimulates PASMC proliferation (Figure 4). In addition to increased PASMC proliferation, decreased PASMC apoptosis may also contribute to the development of pulmonary vascular remodeling in IPAH patients. An early hallmark of cell apoptosis is apoptotic volume decrease (AVD) triggered at least partially by K+ efflux from the cell through K+ channels. Maintenance of a high concentration of cytosolic K+ ([K+]cyt) is not only essential for normal ion homeostasis and cell volume, but also critical for the suppression of cytoplasmic caspases. Therefore, an increased K+ efflux through K+ channels in the plasma membrane causes the apoptotic cell shrinkage or AVD, whereas a decreased K+ channel activity attenuates apoptotic cell shrinkage and maintains sufficient K+ in the cytoplasm to inhibit caspase-mediated apoptosis. We previously demonstrated that the expression and function of Kv channels are significantly reduced, while proliferation is enhanced and apoptosis is inhibited, in IPAH-PASMC in comparison to normal PASMC. This study aims to define the upstream mechanism responsible for the downregulated expression of Kv channels in IPAH-PASMC.
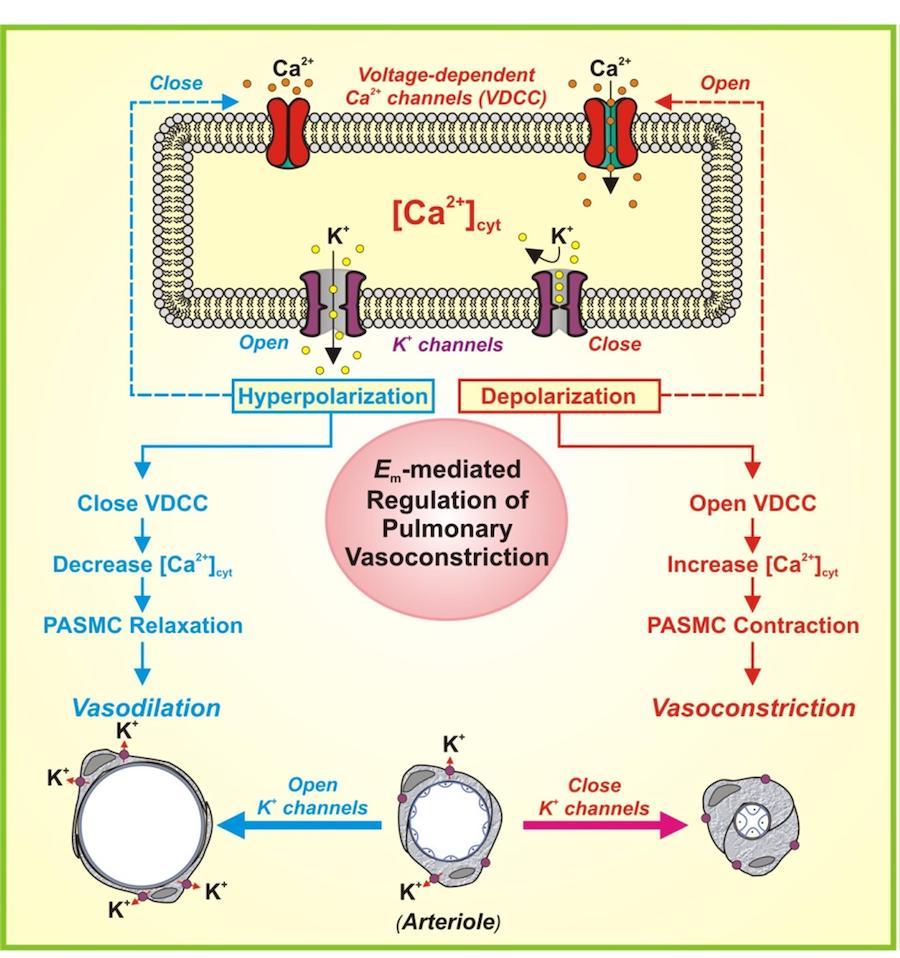
Figure 4. The role of K+ channels in membrane depolarization and pulmonary vasoconstriction. (A) When K+ channels are closed (or K+ channel expression is downregulated), the resulting membrane depolarization opens VDCC, promotes Ca2+ influx, increases [Ca2+]cyt and causes vasoconstriction. When K+ channels are activated (or K+ channel gene expression is upregulated), the resulting membrane hyperpolarization closes VDCC, inhibits agonist-mediated Ca2+ influx and causes vasodilation.
Posttranscriptional processes, particularly altered mRNA turnover and translation, are major mechanisms by which mammalian cells control gene expression. Changes in mRNA stability and translation are regulated by two major types of trans-acting factors that directly interact with the mRNA: non-coding RNAs such as microRNAs (miRNAs) and RNA-binding proteins. miRNAs are short RNAs that bind to complementary sequences in the 3'-untranslated regions (3'-UTR) of target mRNAs to cause mRNA degradation or disrupt mRNA translation, thereby inhibiting the expression of specific mRNA targets. miRNAs are thus posttranscriptional regulators that potentially regulate the level of Kv channel mRNAs in PASMC. We recently identified 5 miRNAs (miR-15a/29a/29b/138/222) that are highly expressed, whereas 9 Kv1 (KCNA) channels (KCNA1-7/10) are significantly downregulated, in IPAH-PASMC compared to normal PASMC. Overexpression of miR-29b, miR-138 or miR-222 in normal PASMC significantly decreases whole-cell Kv currents (IK(V)), whereas inhibition of miR-29b rescues (i.e., significantly enhances) IK(V) in IPAH-PASMC. These data suggest that miR-29b, miR-138 and miR-222 are sufficient to decrease IK(V) in normal PASMC, while miR-29b is necessary for the decreased IK(V) in IPAH-PASMC.
Based on these observations, we hypothesize that selectively upregulated miRNAs are involved in the posttranscriptional inhibition of Kv channels in IPAH-PASMC and the miRNA-mediated Kv channel inhibition contributes to increasing proliferation and decreasing apoptosis in IPAH-PASMC. Downregulation of Kv channels plays an important role in the development of sustained pulmonary vasoconstriction and vascular remodeling in patients with IPAH. The long-term goal of this study is to define the mechanism underlying the inhibition of Kv channels in IPAH-PASMC and to explore the possibility to target miRNA for developing therapeutic approaches for IPAH.
Notch receptors (Notch1-4) are a family of transmembrane proteins composed of functional extracellular, transmembrane, and intracellular domains. There are two types of Notch ligands, Delta-like (Dll1-4) and Jagged (Jag-1/2) in mammals and humans. When Notch ligands, such as Jag-1, in the signal-sending cell bind to the Notch receptor in the signal-receiving cell, it triggers proteolytic cleavages of Notch by the ADAM family of metalloproteases (within the juxtamembrane region) followed by γ-secretase (within the transmembrane domain) resulting in the release of the Notch intracellular domain (NICD) from the membrane to the cytosol. NICD translocates to the nucleus where it interacts with RBP-Jκ and recruits co-activators to turn on expression of Notch target genes. Notch signaling promotes proliferative cascades in cardiomyocytes and smooth muscle cells (SMC). Recent studies show that Notch signaling is required for cellular hypoxic response via interaction between NICD and hypoxia-inducible factor 1a (HIF-1a).
We have ling using DAPT (a γ-secretase inhibitor), while Jag-1 (a Notch ligand) mimics the contractile effect of acute hypoxia to cause pulmonary vasoconstriction in isolated perfused/ventilated mouse lung; b) acute HPV and chronic HPH are significantly inhibited in Trpc6-/-mice compared to the wild-type (WT) littermates; c) acute hypoxia (5-10 min) or short-term Jag-1 treatment (5-10 min) increases NICD and enhances store-operated Ca2+ entry (SOCE) in PASMC; d) chronic hypoxia (2-3 days) or long-term Jag-1 treatment (2-3 days) upregulates TRPC6 and Orai1/2 channels in PASMC; and e) pharmacological blockade of TRPC6 channels significantly attenuated HPH in mice. These data suggest that Notch signaling may functionally activate SOCE channels (e.g., TRPC6 channels) and transcriptionally upregulates TRPC6 channels in PASMC, thereby enhancing the Ca2+signaling cascade required for hypoxia-induced pulmonary vasoconstriction and vascular remodeling.
Based on these data, we hypothesize that hypoxia activates Notch signaling and enhances Notch-mediated functional activation (acute effect) and transcriptional upregulation (chronic effect) of TRPC6 and Orai1/2 channels in PASMC, and subsequently increases Ca2+ influx in PASMC leading to pulmonary vascular remodeling and pulmonary hypertension (Figure 5). We will use combined techniques of molecular and cell biology, patch clamp, fluorescence microscopy, and immunocytochemistry, as well as in vivo animals in this study. Completion of this study will provide important information in understanding the pathogenic mechanisms of HPH associated with abnormal Notch and Ca2+ signaling and in developing novel therapeutic approaches for patients with pulmonary hypertension (PH) associated with hypoxia.
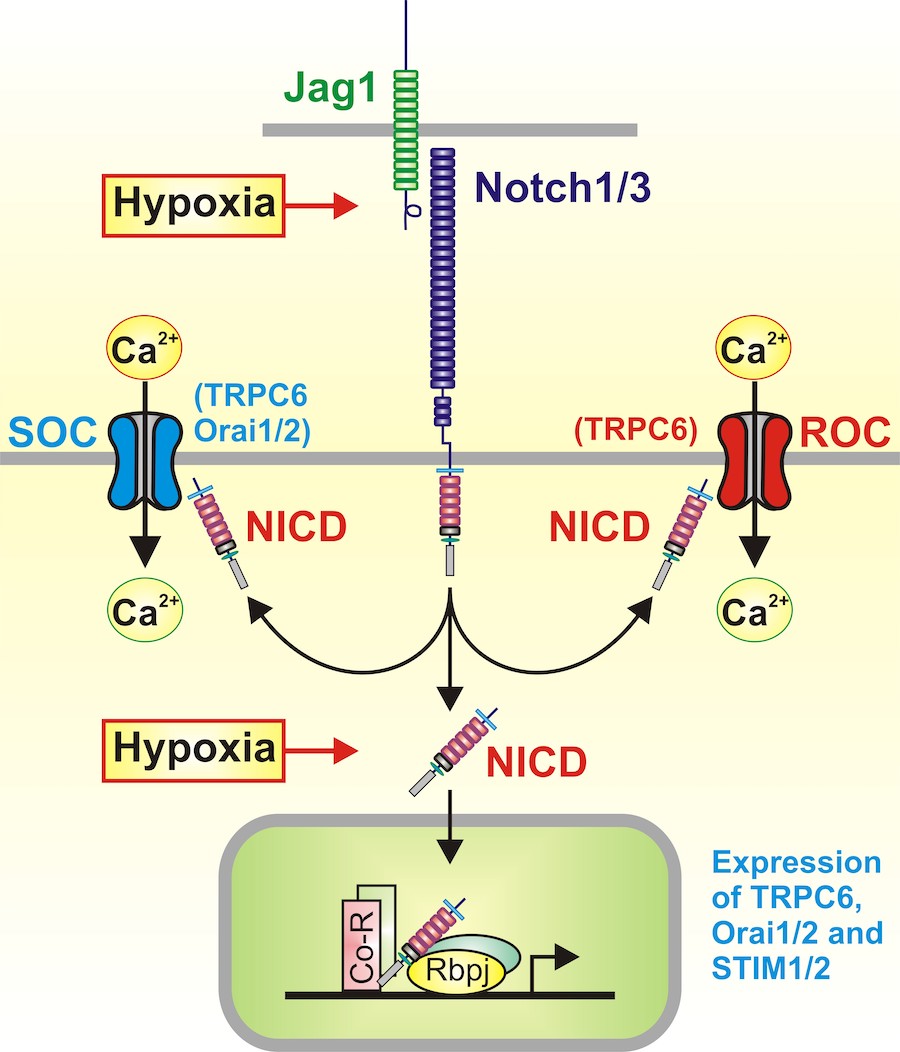
Figure 5. Proposed mechanisms by which NICD (hypoxia-mediated NICD) functionally activates TRPC6- and Orai1/2-formed store-operated (SOC) and receptor-operated (ROC) Ca2+ channels and transcriptionally upregulates SOC/ROC in PASMC.
Lab Techniques Utilized
The Yuan Lab members utilize a multitude of techniques to study the cellular and molecular mechanisms of pulmonary vascular disease and right heart failure. Each member has one or more unique skills that allow him/her to collaborate and examine the various aspects of pathogenic mechanisms and therapeutic interventions of various of pulmonary vascular disease (e.g., idiopathic, thromboembolic and hypoxic pulmonary hypertension). Most of lab members have technical expertise in conventional cellular and molecular biology techniques digital (Fig. 6), such as using RT-PCR and Western Blot to examine mRNA and protein expression level in primary cultured cells from patients and freshly-isolated lung vascular tissues from experimental animals. In collaboration with our clinical colleagues, we conduct cell and molecular biological experiments using lung vascular cells from patients with pulmonary arterial hypertension (via lung transplantation), patients with chronic thromboembolic pulmonary hypertension (via pulmonary endarterectomy and balloon pulmonary angioplasty), patients without pulmonary hypertension (e.g., patients underwent lung lobectomy for cancer), and organ donors (whose lung is not used for clinical treatment)

. General and conventional cell and molecular biological techniques
Using a digital imaging fluorescent microscopy system ;(Fig. 7), we can measure the changes of cytosolic Ca2+ ;concentration ([Ca2+]cyt) due to Ca2+ ;influx and extrusion as well as Ca2+ ;release and uptake in human and animal pulmonary artery smooth muscle and endothelial cells. ; We use primary cultured cells isolated from normal subjects (organ donor) and patients with pulmonary arterial hypertension as well as freshly-dissociated cells from control animals and animals with experimental pulmonary hypertension to analyze [Ca2+]cyt ;and study Ca2+ ;signaling in cell contraction, migration, proliferation, differentiation and apoptosis. These experiments help to understand the pathogenic role of Ca2+-permeable cation channels in the development of pulmonary vasoconstriction, concentric pulmonary vascular remodeling and occlusive vascular lesions, major causes for the elevated pulmonary vascular resistance in patients with pulmonary arterial hypertension and animals with experimental pulmonary hypertension.
Figure 7. Digital imaging fluorescence microscopy.
Another electrophysiological approach utilized by the lab is called patch clamp technique (Fig. 8). The patch clamp technique allows for the study of single or multiple ion channels in a single cell. We can measure the whole-cell ionic current with the whole-cell mode and single-channel current in the cell-attached patch, outside-out patch and inside-out patch. We can also use the technique to measure membrane potential and action potentials in excitable cells using the current-clamping mode. Patch clamping is a standard electrophysiological technique for fundamental studies of ion channels and membrane receptors, cell excitability and excitation-contraction coupling, which is widely implemented in the discovery of drugs that affect these proteins for cardiopulmonary and vascular diseases.
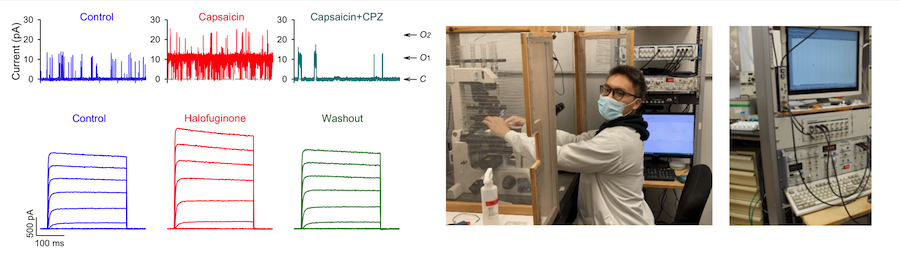
Figure 8. Patch clamp techniques.
This lab utilizes different animal (rat and mouse) models (Fig. 9) of pulmonary hypertension to investigate the pathogenic mechanisms and therapeutic interventions of pulmonary hypertension. We use rats with monocrotaline (MCT)-induced pulmonary hypertension (MCT-PH) and mice with sugen/hypoxia-induced pulmonary hypertension (Su/Hx-PH), as models for severe pulmonary hypertension, and rats or mice with chronic hypoxia-induced pulmonary hypertension (HPH), as models for mild pulmonary hypertension. We also use globule, conditional and inducible knockout mice as well as transgenic mice for in vivo experiments. Our lab members conduct right heart catheterization (RHC) in mice and rats (Fig. 9) to measure right ventricle systolic pressure, as surrogate measure for pulmonary arterial systolic pressure, to study changes of pulmonary hemodynamics and right ventricular function in intact animals with or without therapeutic interventions and in wild-type and knock-out mice.
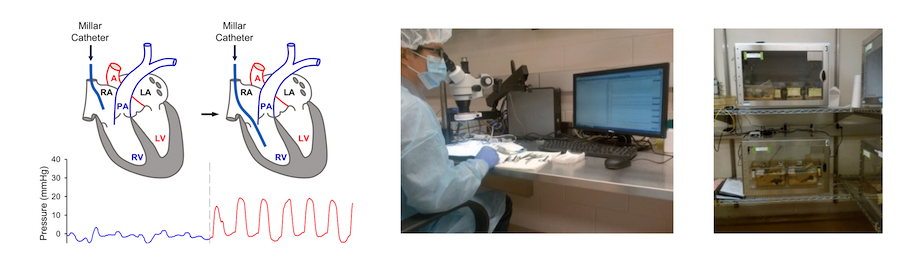
Figure 9. Animal models of pulmonary hypertension and right heart catheterization in mice.
We also conduct ex vivo experiments using isolated perfused/ventilated mouse lung (Fig. 10) to study pulmonary vascular reactivity and mouse lung angiogram (Fig. 10) to study pulmonary vascular remodeling. Isolated perfused/ventilated mouse lung preparation is also used to study cellular mechanisms of alveolar hypoxic pulmonary vasoconstriction, an important physiological mechanism involved in assuring gas exchange and blood oxygenation. Ex vivo pulmonary angiography is used to study pulmonary vascular structural changes during the development of experimental pulmonary hypertension and identify new drugs that may result in the regression of remodeled pulmonary arteries and reversal (or partial reversal) of experimental pulmonary hypertension.
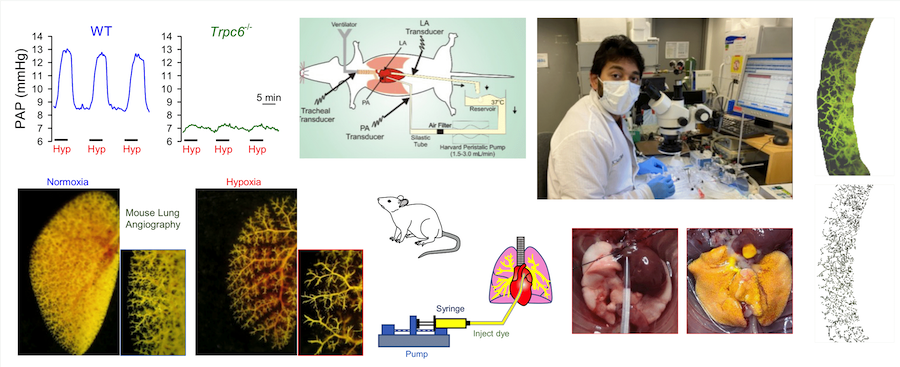
Figure 10. Isolated perfused/ventilated mouse lung and ex vivo lung angiography